Error Tolerant Search
When the results of an MS/MS Ions Search of an LC-MS/MS dataset are reviewed, there will often
be a number of spectra that remain unmatched. Assuming that a given MS/MS spectrum contains
adequate information, i.e. a reasonable number of fragment ion peaks at usable signal to noise,
possible reasons for this failure include:
- Underestimated mass measurement error
- Incorrect determination of precursor charge
- Enzyme non-specificity
- Unsuspected chemical & post-translational modifications
- Peptide sequence not in the database
If mass measurement error has been underestimated, this should be apparent from the graphs
showing the differences between the calculated and measured mass values in the Peptide View
and Protein View reports.
Incorrect determination of precursor charge has to be dealt with during peak detection. If it is not
possible to determine the precursor charge reliably, then one option is to generate peak lists
for all probable charge states.
The Mascot Error Tolerant Search addresses the final three difficulties by searching
selected database entries with relaxed enzyme specificity, while iterating through a comprehensive
list of chemical and post-translational modifications, together with a residue
substitution matrix.
There are two ways to perform an error tolerant search. The preferred method is to check the
error tolerant checkbox on the search form, which leads to an automatic, second pass search.
There is also a manual procedure, in which the user selects the proteins that will go forward
for the second pass search. This was an earlier implementation, and is retained mainly
for compatibility with existing workflows and third party software.
Note that both methods are only applicable to MS/MS data; it is not possible to perform an error tolerant
peptide mass fingerprint. For a truly unknown modification, or a sequence variation
of more than a single base or residue, the error tolerant
sequence tag is worth investigating.
An automatic error tolerant search is performed by choosing the error tolerant checkbox on
the search form. A standard, first pass search is performed using the search parameters specified
in the form.
From the results of the first pass search, all of the database entries that contain one or
more peptide matches with scores at or above the homology threshold, (or identity threshold
if there is no homology threshold), are selected for an error tolerant,
second pass search. At the completion
of the second pass search, as single report is generated, combining the results from both passes.
During the error tolerant, second pass search:
- The selected enzyme becomes semi-specific, (that is, only one end of a peptide needs
to match the cleavage specificity), and the value of the missed cleavage parameter
is increased by 1
- The complete list of modifications is tested, serially
- For a protein, the set of substitutions that can arise from single base
substitutions is tested. For a nucleic acid sequence, all single base insertions,
deletions, and substitutions are tested.
- Only one of the above is allowed per peptide. That is, an individual peptide
can be semi-specific OR have one unsuspected modification OR have
one primary sequence mutation.
- If the modified and unmodified peptides are both within the precursor mass tolerance
window, the modification is rejected. This eliminates modifications that are
meaningless given the estimated mass error, like Q->K, in most cases.
The following constraints apply to the standard, first pass search:
- Enzyme must be fully specific
- A reduced ceiling on the number of variable modifications, (default is 2, but this can be changed globally
in mascot.dat or for a user group in Mascot security)
- Cannot be combined with an automatic decoy database search
- Cannot be combined with quantitation
- Search cannot include error tolerant sequence tag
Database entries are selected from the results report of a standard search. Check the
Error tolerant checkbox, near the Search selected button, and choose one or more
proteins to be included in the search. (On the public web
site, a maximum of 3 proteins can be chosen). Clicking on the Search selected button loads a modified
search form, from which you can change many of the search parameters. Cleavage agent defaults to None,
though an enzyme can be chosen if desired.
During the error tolerant search:
- The complete list of modifications is tested, serially
- For a protein, the set of substitutions that can arise from single base
substitutions is tested. For a nucleic acid sequence, all single base insertions,
deletions, and substitutions are tested.
The manual error tolerant search should only be used in
exceptional cases. One reason is that, because enzyme specificity is dropped entirely, and modifications can be
combined with non-specificity, and the number of database entries tends to be fewer,
the level of "junk" matches in the manual search will be higher
than in the automatic search. Another reason is that, in the automatic search, the results from both
passes are saved to the result file, which provides
greater reporting flexibility. For example, you can choose to show or hide the additional, error
tolerant matches. The combined report also reduces compatibility problems for applications
that read Mascot result files.
It is important to recognise that only the matches from the
standard, first pass search provide evidence for the identity of a protein. The
additional matches found in the error tolerant, second pass search are valuable
because they are the most likely
assignments of the spectra. Occasionally, an additional match will provide useful
biological information, such as disinguishing between two isoforms. If the same
modification shows up many times, this may indicate an experimental artefact that
needs to be eliminated or, at least, selected as a variable modification for
standard searches.
Nevertheless, these additional matches have been obtained by selecting a small number
of database entries and beating them into submission with non-specificity, substitutions
and a long list of modifications. This makes it difficult to apply any meaningful
measure of statistical significance. So, in the result report, the error tolerant matches
are treated differently from the standard matches:
- They do not contribute to the protein score. (If the query also has
a lower scoring match to the same protein in the first pass search,
this contributes, so that the protein scores are identical to those
seen in the standard report.
- Significance thresholds are not reported
- Expect values are not reported
- Must have scores of at least the identity threshold
for the query in the first pass search
- Must have scores in excess of the highest scoring match
to the query in the first pass search
(Item 1 only applies to the combined report, obtained from an automatic error tolerant search.
If you perform a manual error tolerant search, the report will show protein scores derived
from all the matches listed.)
For example, click on this thumbnail image to load an example of the results from an error tolerant
search in a new browser window. Scroll down to hit 1, Alkaline phosphatase.
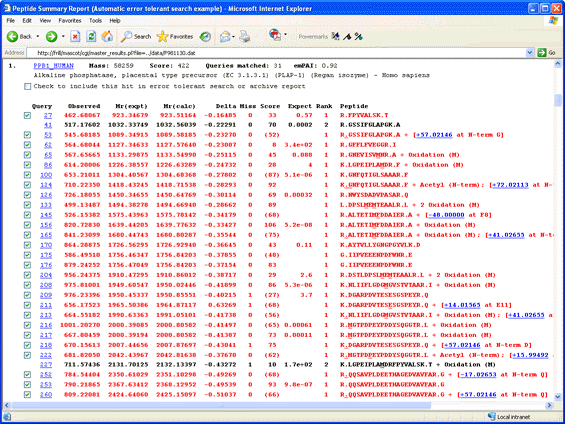
The additional. error tolerant matches can be recognised easily as the ones with gaps
in the expect value column. In some cases, the additional match is the result of non-specific cleavage,
such as queries 133 and 176. If the error tolerant match was found by introducing a modification
or a sequence change, the mass delta and its location are given at the end of the row. When the mouse rests over the mass
delta hyperlink, all the known assignments of this delta are displayed in a pop-up. Take a look at
query 218. The mass tolerance for this search was fairly wide, ±0.8 Da, so the observed mass difference could
correspond to either carbamidomethylation or carboxymethylation at the N-terminus. Since this sample was
alkylated with iodoacetamide, we would choose carbamidomethylation as the more likely suspect, especially as
this brings the error on the precursor mass into line with the general trend, whereas carboxymethylation would
give an error of +0.6 Da. The assignment to carbamidomethylation is also very
believable, because this is a known artefact of over-alkylation. The same modification is found for
queries 53 and 260.
Another easily believable assignment is pyro-Glu for the match to query 252.
In other cases, the match may be good, but the assignment is not believable. For example, look at
query 145, which has a mass difference of -48.0 Da, assigned to a substitution, F->V. This is
not feasible because the native peptide is matched strongly by the next two queries. Figuring out
a reasonable assignment for some matches can require a bit of detective work. In this case,
notice that the next two matches show the Met as being oxidised. If we hypothesise that the Met in the
match to query 145 is also oxidised, this would take the mass difference to -64 Da. It is
well known that oxidised Met loses 64 Da, (methanesulfenic acid),
during CID. The most likely explanation for this match is that there was prompt loss
of 64 Da from oxidised Met, possibly due to in-source collision.
The other place to look for clues is the list of alternative matches that
is displayed when the mouse cursor rests over the query number. For example, scroll down to hit 2,
bovine trypsin, and look at the pop-up for query 251. Several possible modification states
give very similar scores:
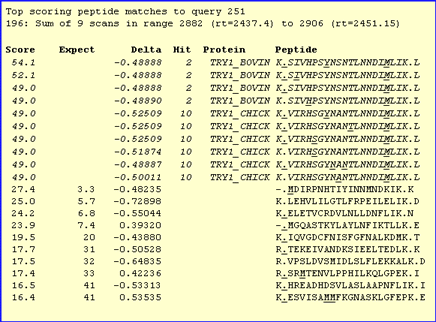
Click on the hyperlink to generate a Peptide View report for this match. There are links at the bottom
of the report to switch between the alternative matches. The top match requires an unlikely modification, Amino (Y). The
second is no better, requiring a substitution. The third is our old friend Carbamidomethyl (N-term). Even
though this has a slightly lower score, it is a rather more credible explanation for the match.
The list of modifications used by Mascot is taken directly from the
Unimod database. For further
details of individual modifications, please refer to Unimod.
Note that only a small sub-set of modifications is displayed by default in the
Mascot search form. If you want to see the complete list, you must go to the
search form defaults page and tick the
checkbox for Show all mods.
In an Error Tolerant search, all the entries on the modifications list are tested serially,
and all permutations of each individual modification are tested.
For example, if a modification affects serine, and a peptide contains three serines, but has a molecular
mass consistent with just two modifications, there are 3 permutations to be tested (110,101,011).
This differs from the behaviour for any variable modifications explicitly specified in the search
form, when all permutations and combinations of the selected modifications are tested. Specifying more
than a handful of variable modifications leads to a drastic loss of discrimination, because the number
of permutations and combinations increases geometrically with the total fractional abundance of modifiable residues.
Variations in the primary sequence generally result from variations in the DNA sequence.
These may be DNA sequencing errors, they may be mutations or polymorphisms,
or they may be more extensive evolutionary changes, because the database entry is not the authentic
protein, but a related sequence from a different species.
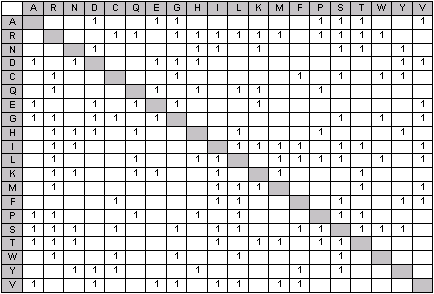
Back translation of AA sequence to NA, expansion of all possible single base substitutions,
and translation back to AA yields the substitution matrix shown below. This contains
150 substitutions, a factor of 2.5 smaller than all possible AA substitutions, and these are
the substitutions that are included in Unimod and are used by
Mascot in an error tolerant search of a protein database.
When searching a nucleic acid database, single base deletions and insertions can be tested in addition
to substitutions. The consequences of deletions and insertions cannot be tested for a protein database
because they cause a frame shift, which completely changes the amino acid sequence from that point onwards.
|