Accuracy & Resolution
Types of Mass Measurement
When low molecular weight samples are being analysed using
relatively low resolution mass spectrometers, it is common to
work with "nominal" mass values, calculated from integer
atomic weights. That is, H=1, C=12, N=14, O=16, etc. Nominal
mass is rarely used in peptide and protein work because the cumulative
error of approximating atomic weights with integers becomes unacceptable.
The presence of isotopes at their natural abundances makes
it essential to define whether an experimental mass value is
an "average" value, equivalent to taking the centroid
of the complete isotopic envelope, or a "monoisotopic"
value, the mass of the first peak of the isotope distribution.
For peptides and proteins, the difference between an average
and a monoisotopic weight is approximately 0.06%. This is a significant
difference when even the most modest instruments are capable
of measuring the mass of a small peptide with an accuracy of a
fraction of a Dalton. For example, peptide HLKTEAEMK has an average
molecular weight of 1086.28 and a monoisotopic weight of 1085.55.
At a mass resolution of 5000, the isotopic envelope has this
appearance:
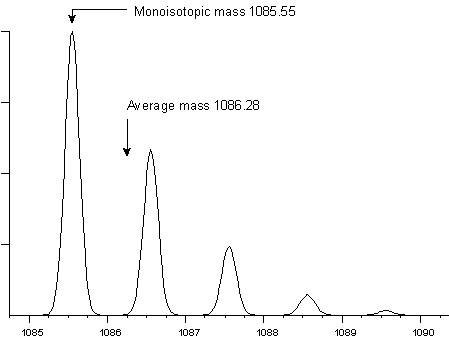
Mass resolution is the dimensionless ratio of the mass of
the peak divided by its width. Usually, the peak width is taken
as the full width at half maximum intensity, (fwhm). However,
this definition of peak width is only a convention, and you may
also encounter data acquired on magnetic sector instruments where
the resolution has been calculated using the peak width at 5%
maximum intensity.
To measure a monoisotopic molecular weight requires (i) sufficient
mass resolution to resolve the the isotopic distribution (ii)
sufficient signal to noise to be able to identify the first peak
of the envelope with confidence. For a small peptide, the first
peak (often referred to as the 12C peak) is also the
most intense peak. This is not the case for larger molecules.
The following two examples show the isotopic envelopes for a
small protein (insulin) and a larger protein (BSA):
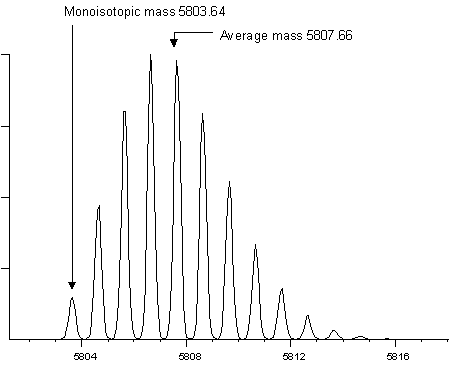
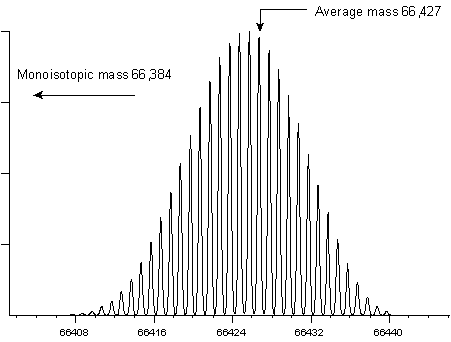
It would be extremely difficult to measure a monoisotopic
mass for BSA, and it is far from routine to measure a monoisotopic
mass for insulin. In practice, most instruments report monoisotopic
molecular weights up to a certain cut-off point. Above this cut-off,
isotopic envelopes are centroided as a whole to provide average
mass values.
(The isotopic distributions shown on this page were calculated
using Mike Senko's IsoPro
program, based on Jim Yergey's algorithm, [Yergey,
1983].)
Mass Accuracy
A monoisotopic mass can be measured as accurately as the instrumentation
allows, as long as the monoisotopic peak has been correctly
identified. If the wrong peak has been chosen, the mass value
will be in error by one or more Daltons, how ever many decimal
places are present. Such mistakes provide a nice demonstration
of the difference between accuracy and precision.
Even a monoisotopic mass peak has a finite width, resulting
from imperfections in real-life mass analysers. This means that
some level of mass measurement error will be caused by statistical
fluctuations in the ion population being measured, but such errors
are small compared with random and systematic errors from other
sources.
This is not the case for an average mass, where the error
in estimating the precise centroid of the full isotopic envelope
may dominate. Isotope distributions are not symmetric, so it
is essential to calculate a centroid, rather than just taking
the apex of the distribution. The accuracy of a centroid depends
on the precise measurement of the ion current at a sufficient
number of points to define the isotopic envelope precisely. Ion
statistics, overlapping distributions, detector saturation, and
a number of other factors can distort relative intensities across
the distribution and so contribute to error in the average mass
value.
In addition to errors associated with instrumentation, the
average mass of a molecule is subject to variations in the isotopic
abundances of its constituent elements. Natural isotopic
abundances depend on the source of the material. For this
reason, the average atomic
weight of carbon, 12.0107±0.0008,
can only be quoted with fairly limited precision. Fortunately,
variations in the 13C/12C ratio within
specific isotopic reservoirs are much smaller. For example, terrestrial
plants which utilise the 3-carbon or C3 photosynthetic
pathway have an average 13C content of -26.6±3
per mil, equivalent to an uncertainty in the atomic weight for
carbon of only ±0.00003 [Mendelsohn,
1986]. This means that, as long as the mass scale of
the instrument is calibrated using molecules derived from the
same isotopic reservoir as the analytes, variations in isotopic
abundance can be neglected.
Resolution
The mass resolution achievable by a mass spectrometer depends
on both the type of analyser and the experimental conditions.
Simple MALDI-TOF instruments may only achieve unit mass resolution
over a limited mass range. High performance FTMS systems can
achieve resolving powers of several hundred thousand.
However, where two species have isotopic envelopes which significantly
overlap, deconvolution of the two envelopes is never a practical
proposition, no matter how much resolution is available.
The factor which complicates any general discussion of resolution
optimisation is that some types of mass analyser have a trade-off
between resolution and sensitivity, while others do not. Where
a monoisotopic peak for a single molecular species can be resolved,
mass accuracy tends to follow resolution. This is because the
narrower the peak, the less the significance of errors due to
variations in the peak shape. So, if unit mass resolution is
possible, then the more resolution the better ... unless
there is a sensitivity trade-off.
If unit mass resolution is not possible, then there is little
benefit to exceeding the instrument resolution at which the isotopic
envelope can be defined without significant broadening. For example,
the following figure shows the molecular ion of glucagon at resolutions
of 1000 (blue), 3000 (red), 10,000 (green) and 30,000 (black).
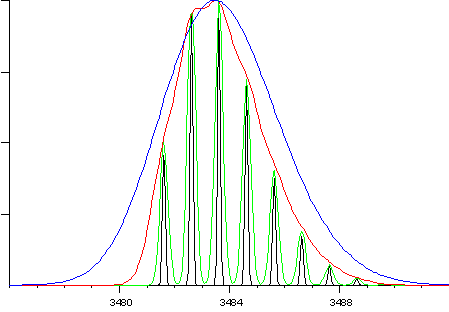
For an average mass measurement, and where there is no trade-off
between sensitivity and resolution, the accuracy at 3000 resolution
(red) will be just as good as at higher resolution. On an instrument
where a trade-off exists, using a resolution greater than 3000
is very likely to degrade mass accuracy.
The Limits of Accuracy
Leucine and isoleucine are isobaric. Using mass spectrometry
alone, they can only be differentiated by observing side chain
fragmentation in MS/MS following high energy collisions. There
are two other common cases where pairs of residues have the same
nominal mass: glutamine and lysine (128), and Phenylalanine and
Met-ox (147). To distinguish these residues in MS/MS spectra
by mass difference alone is rarely a practical proposition.
|